The use of next‐generation sequencers and advanced genotyping technologies has propelled the field of plant genomics in model crops and plants and enhanced the discovery of hidden bridges between genotypes and phenotypes. The newly generated reference sequences of unstudied minor plants can be annotated by the knowledge of model plants via translational genomics approaches. Here, we reviewed the strategies of translational genomics and suggested perspectives on the current databases of genomic resources and the database structures of translated information on the new genome. As a draft picture of phenotypic annotation, translational genomics on newly sequenced plants will provide valuable assistance for breeders and researchers who are interested in genetic studies.
Published papers

Translational genomics for plant breeding with the genome sequence explosion

Genome-wide DNA methylation profile in mungbean
DNA methylation on cytosine residues is known to affect gene expression and is potentially responsible for the phenotypic variations among different crop cultivars. Here, we present the whole-genome DNA methylation profiles and assess the potential effects of single nucleotide polymorphisms (SNPs) for two mungbean cultivars, Sunhwanogdu (VC1973A) and Kyunggijaerae#5 (V2984). By measuring the DNA methylation levels in leaf tissue with the bisulfite sequencing (BSseq) approach, we show both the frequencies of the various types of DNA methylation and the distribution of weighted gene methylation levels. SNPs that cause nucleotide changes from/to CHH – where C is cytosine and H is any other nucleotide – were found to affect DNA methylation status in VC1973A and V2984. In order to better understand the correlation between gene expression and DNA methylation levels, we surveyed gene expression in leaf tissues of VC1973A and V2984 using RNAseq. Transcript expressions of paralogous genes were controlled by DNA methylation within the VC1973A genome. Moreover, genes that were differentially expressed between the two cultivars showed distinct DNA methylation patterns. Our mungbean genome-wide methylation profiles will be valuable resources for understanding the phenotypic variations between different cultivars, as well as for molecular breeding.

Resequencing of Capsicum annuum parental lines (YCM334 and Taean) for the genetic analysis of bacterial wilt resistance
Bacterial wilt (BW) is a widespread plant disease that affects a broad range of dicot and monocot hosts and is particularly harmful for solanaceous plants, such as pepper, tomato, and eggplant. The pathogen responsible for BW is the soil-borne bacterium, Ralstonia solanacearum, which can adapt to diverse temperature conditions and is found in climates ranging from tropical to temperate. Resistance to BW has been detected in some pepper plant lines; however, the genomic loci and alleles that mediate this are poorly studied in this species. We resequenced the pepper cultivars YCM344 and Taean, which are parental recombinant inbred lines (RIL) that display differential resistance phenotypes against BW, with YCM344 being highly resistant to infection with this pathogen. We identified novel single nucleotide polymorphisms (SNPs) and insertions/deletions (Indels) that are only present in both parental lines, as compared to the reference genome and further determined variations that...

Genome sequence of mungbean and insights into evolution within <i>Vigna</i> species
Mungbean is a fast-growing and warm-season legume crop, cultivated mainly in Asia. Here, the authors sequence the genomes of both wild and domesticated mungbean varieties and, together with detailed transcriptome data, provide insight into mungbean domestication, polyploidization and speciation.
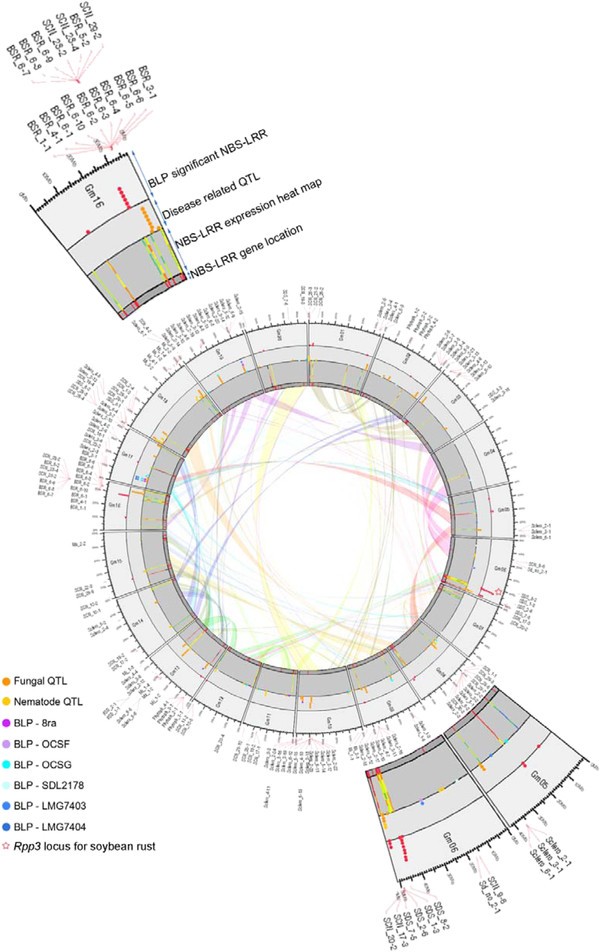
Genome-wide mapping of NBS-LRR genes and their association with disease resistance in soybean
R genes are a key component of genetic interactions between plants and biotrophic bacteria and are known to regulate resistance against bacterial invasion. The most common R proteins contain a nucleotide-binding site and a leucine-rich repeat (NBS-LRR) domain. Some NBS-LRR genes in the soybean genome have also been reported to function in disease resistance. In this study, the number of NBS-LRR genes was found to correlate with the number of disease resistance quantitative trait loci (QTL) that flank these genes in each chromosome. NBS-LRR genes co-localized with disease resistance QTL. The study also addressed the functional redundancy of disease resistance on recently duplicated regions that harbor NBS-LRR genes and NBS-LRR gene expression in the bacterial leaf pustule (BLP)-induced soybean transcriptome. A total of 319 genes were determined to be putative NBS-LRR genes in the soybean genome. The number of NBS-LRR genes on each chromosome was highly correlated with the number of...
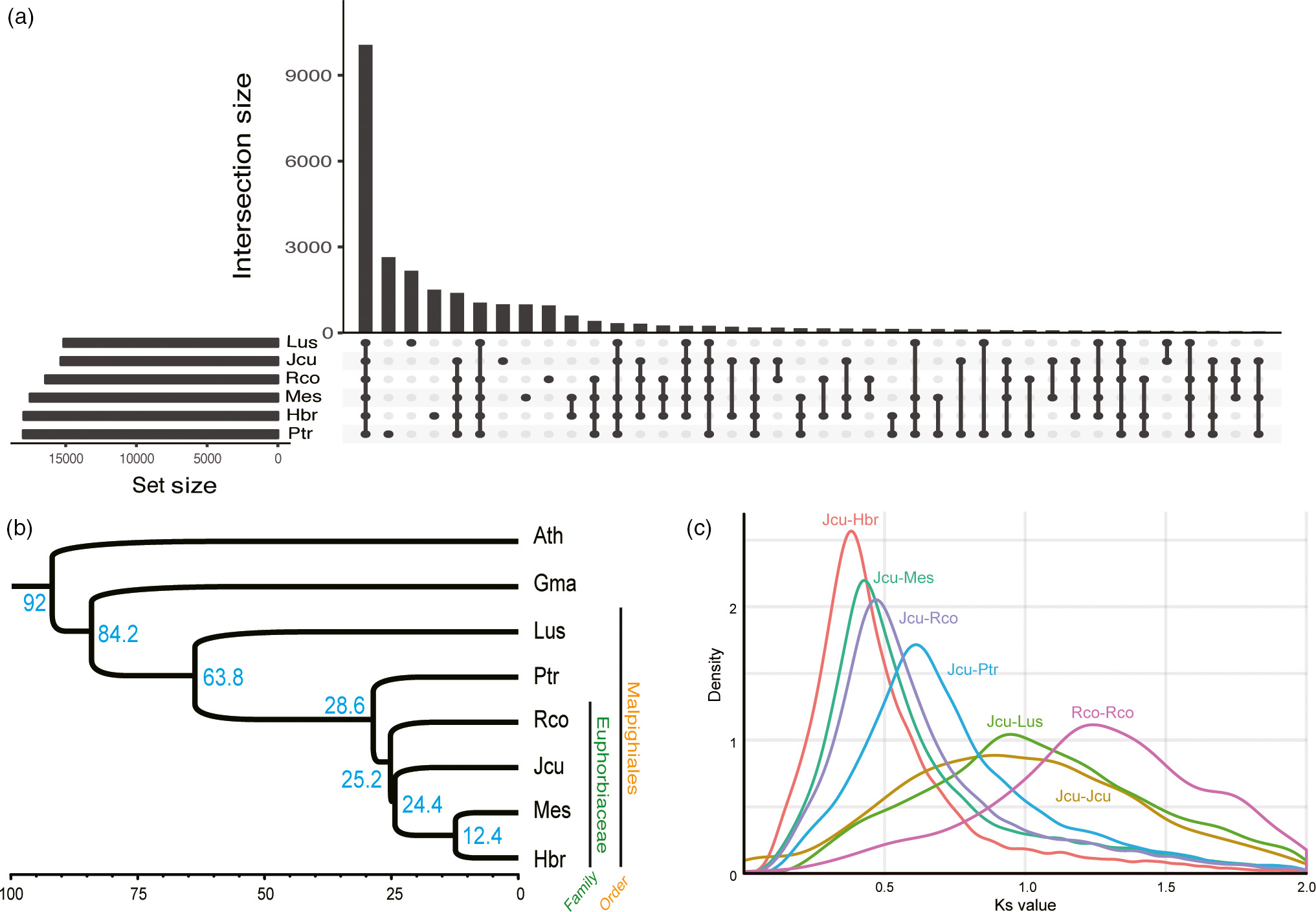
Genome sequence of Jatropha curcas L., a non‐edible biodiesel plant, provides a resource to improve seed‐related traits
Jatropha curcas (physic nut), a non‐edible oilseed crop, represents one of the most promising alternative energy sources due to its high seed oil content, rapid growth and adaptability to various environments. We report ~339 Mbp draft whole genome sequence of J. curcas var. Chai Nat using both the PacBio and Illumina sequencing platforms. We identified and categorized differentially expressed genes related to biosynthesis of lipid and toxic compound among four stages of seed development. Triacylglycerol (TAG), the major component of seed storage oil, is mainly synthesized by phospholipid:diacylglycerol acyltransferase in Jatropha, and continuous high expression of homologs of oleosin over seed development contributes to accumulation of high level of oil in kernels by preventing the breakdown of TAG. A physical cluster of genes for diterpenoid biosynthetic enzymes, including casbene synthases highly responsible for a toxic compound, phorbol ester, in seed cake, was syntenically highly conserved between Jatropha and castor bean. Transcriptomic analysis of female and male flowers revealed the up‐regulation of a dozen family of TFs in female flower. Additionally, we constructed a robust species tree enabling estimation of divergence times among nine Jatropha species and five commercial crops in Malpighiales order. Our results will help researchers and breeders increase energy efficiency of this important oil seed crop by improving yield and oil content, and eliminating toxic compound in seed cake for animal feed.
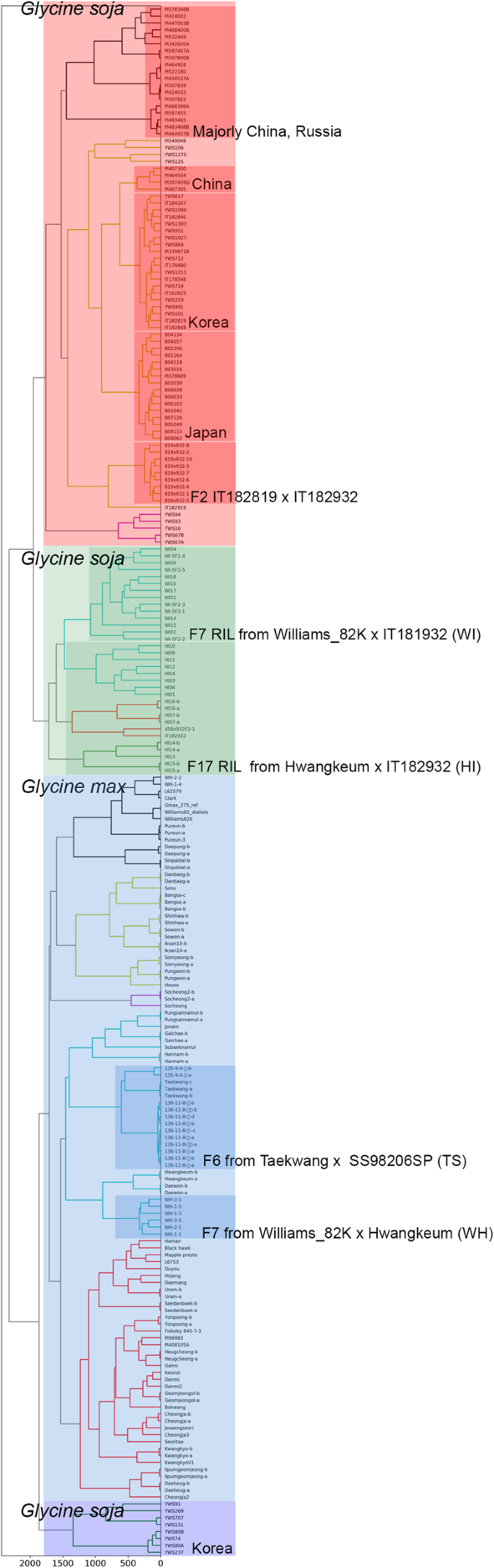
Soybean-VCF2Genomes: a database to identify the closest accession in soybean germplasm collection
The development of next generation sequencer (NGS) and the analytical methods allowed the researchers to profile their samples more precisely and easier than before. Especially for agriculture, the certification of the genomic background of their plant materials would be important for the reliability of seed market and stable yield as well as for quarantine procedure. However, the analysis of NGS data is still difficult for non-computational researchers or breeders to verify their samples because majority of current softwares for NGS analysis require users to access unfamiliar Linux environment. Here, we developed a web-application, “Soybean-VCF2Genomes”, http://pgl.gnu.ac.kr/soy_vcf2genome/ to map single sample variant call format (VCF) file against known soybean germplasm collection for identification of the closest soybean accession. Based on principal component analysis (PCA), we simplified genotype matrix for lowering computational burden while maintaining accurate clustering....

Database: web application for visualization of the cumulated RNAseq data against the salicylic acid (SA) and methyl jasmonate (MeJA) treatment of Arabidopsis thaliana
Plants have adapted to survive under adverse conditions or exploit favorable conditions in response to their environment as sessile creatures. In a way of plant adaptation, plant hormones have been evolved to efficiently use limited resources. Plant hormones including auxin, jasmonic acid, salicylic acid, and ethylene have been studied to reveal their role in plant adaptation against their environment by phenotypic observation with experimental design such as mutation on hormone receptors and treatment / non-treatment of plant hormones along with other environmental conditions. With the development of Next Generation Sequencing (NGS) technology, it became possible to score the total gene expression of the sampled plants and estimate the degree of effect of plant hormones in gene expression. This allowed us to infer the signaling pathway through plant hormones, which greatly stimulated the study of functional genomics using mutants. Due to the continued development of NGS technology and analytical techniques, many...

Draft genome sequence of adzuki bean, Vigna angularis
Adzuki bean (Vigna angularis var. angularis) is a dietary legume crop in East Asia. The presumed progenitor (Vigna angularis var. nipponensis) is widely found in East Asia, suggesting speciation and domestication in these temperate climate regions. Here, we report a draft genome sequence of adzuki bean. The genome assembly covers 75% of the estimated genome and was mapped to 11 pseudo-chromosomes. Gene prediction revealed 26,857 high confidence protein-coding genes evidenced by RNAseq of different tissues. Comparative gene expression analysis with V. radiata showed that the tissue specificity of orthologous genes was highly conserved. Additional re-sequencing of wild adzuki bean, V. angularis var. nipponensis, and V. nepalensis, was performed to analyze the variations between cultivated and wild adzuki bean. The determined divergence time of adzuki bean and the wild species predated archaeology-based domestication time. The present genome assembly will accelerate the genomics-assisted breeding of adzuki bean.
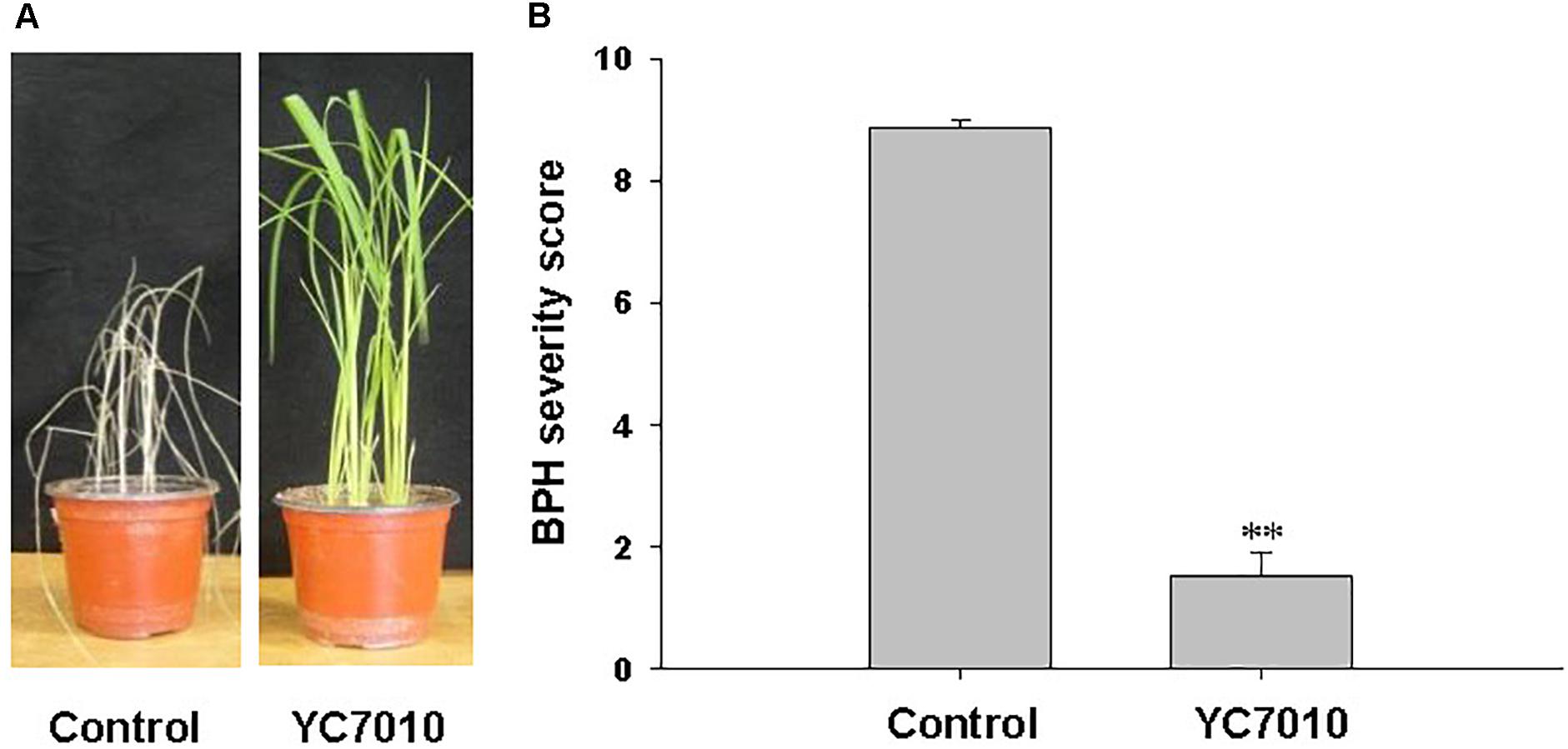
Defenses Against Brown Planthopper Through Transcriptomic and Metabolic Changes in Rice
Brown planthopper (BPH; Nilaparvata lugens Stål) is one of the most serious insect pests, which reduce rice yield remarkably in many rice-growing areas. A few plant growth-promoting rhizobacteria induce systemic resistance against herbivorous insects. Here we show that root drenching of rice seedlings with an endophytic strain Bacillus velezensis YC7010 enhanced defenses against BPH. Based on high-throughput transcriptome analysis, systemic resistance against BPH was induced by B. velezensis YC7010 via salicylic acid (SA)- and jasmonic acid (JA)-dependent pathways. Increased leaf contents of secondary metabolites, tricin and C-glycosyl flavone and cell-wall contents of lignin and cellulose were the key defense mechanisms inducing resistance against BPH during the three-way interaction. This study shows for the first time that chemical changes and strengthening of physical barriers play important roles simultaneously in plant defense against BPH in rice by the endophytic bacteria. This defense was induced by lipopeptides including a novel bacillopeptin X.